
真野 叶子1、安藤 香奈絵2
1名古屋大学 環境医学研究所 病態神経科学分野
2東京都立大学 理学部 生命科学科
はじめに
ミトコンドリアは古くから研究されている馴染み深い細胞小器官であるにもかかわらず、未だに新発見が相次いでいる。私たちの脳は膨大なエネルギーを必要とし、エネルギー生産をはじめ多くをミトコンドリアに依存している。私たちの脳の神経細胞ほとんどは、生涯入れ替わることなく個体と共に老化する。しかし、加齢による神経細胞内のミトコンドリアの変化や、その個体老化への影響については、未だに不明な点が多い。本稿では、神経細胞の老化と疾患におけるミトコンドリアの役割から、神経変性疾患治療の今後の展望を考えたい。
神経細胞の機能維持におけるミトコンドリアの役割
ミトコンドリアの役割というと真っ先に思い浮かぶのがエネルギー産生である。神経細胞はエネルギー源として糖に依存しており、加齢によるATPの減少は神経細胞の老化の原因となる(1)。糖代謝の過程でミトコンドリアはTCA回路から酸化的リン酸化によりATPを産生する。酸化的リン酸化はミトコンドリア内膜にある電子伝達系で行われる。複合体IとIIにより電子を供給し、複合体III, IV, Vと反応が続くことで、最終的に糖1分子から36のATPが産生される。ミトコンドリア呼吸鎖複合体の変異は「ミトコンドリア病」と総称される遺伝性疾患の原因であり、それらの多くで重篤な神経症状がみられる。ミトコンドリア病の多くは若齢で発症するが、パーキンソン病のような加齢依存性神経変性疾患を引き起こす変異もある(2)。
さらに、ミトコンドリアは脂質代謝にも重要な役割を持つ。神経細胞の機能と老化に脂質は大きく関わるが、脳での脂肪滴の蓄積は、老化や神経変性疾患の発症機構に関わるとして注目されている。ミトコンドリアでのATP産生の副産物の活性酸素種(Reactive Oxygen Species (ROS))は細胞にダメージを与えるが、神経細胞はROSが上昇すると酸化された脂質を放出し、これをグリア細胞が受け取って脂肪滴を形成する(3)。ミトコンドリア内で脂質のβ酸化を行うことはよく知られているが、近年ミトコンドリアが脂肪滴と接触し、その生成に関わることも報告された(4)。ミトコンドリアは脂肪滴を介した脳の老化にも関わる可能性がある。
また近年、ミトコンドリアが細胞内のタンパク質恒常性に寄与することが明らかになってきた。ミトコンドリアは自身のタンパク質を制御するため、ミトコンドリア内部にタンパク質分解酵素や、シャペロン、さらには外膜にもユビキチン化酵素をもつ。酵母ではミトコンドリアが細胞質で凝集したタンパク質を内部に取り込み、自身のプロテアーゼで分解することが報告された(5)。さらに私たちの研究から、神経細胞内のプロテアソームやオートファジーといった細胞質のタンパク質分解系の制御にミトコンドリアが寄与していることが明らかになった(6)。多くの加齢依存性神経変性疾患は特定のタンパク質の凝集や蓄積が原因になることから、ミトコンドリアによる細胞質タンパク質分解制御の低下が発症機構に関わる可能性がある。
老化による神経細胞内のミトコンドリア変化
ミトコンドリアは細胞内のATP需要を満たすため、細胞の隅々まで能動的に輸送される。核のある細胞体から長く伸びた軸索、樹状突起と極性に富んだ構造を持つ神経細胞ではこの輸送が特に重要である。ミトコンドリアの能動輸送のメカニズムの解明にはショウジョウバエの遺伝学が大きな貢献をしている。視覚を失う変異体のスクリーンから同定されたmiltonは、ミトコンドリアとキネシンモーターを繋ぐアダプタータンパク質をコードしていることがわかった(7)。miltonと結合して共にアダプターを構成するMiroはミトコンドリアの外膜に存在し、カルシウムに応答してその構造を変化させることでミトコンドリアをキネシンから離す(8)。つまり、ミトコンドリアはmiltonとキネシンから成る電車に乗り、微小管というレールの上を走っているが、カルシウムイオンによる停止信号により必要な場所で下車できるというわけである。
ミトコンドリアの軸索輸送は加齢と共に低下する(9)。そして、このミトコンドリア輸送の低下が神経細胞の老化を促進していると考えられる。例えばmiltonを神経細胞でノックダウンしたショウジョウバエでは全体のミトコンドリアの量は変わらないものの、輸送が停止し軸索ではミトコンドリアが減少する。このショウジョウバエは、羽化直後は野生型と違いが見られないが、老化に伴い変化がでてくる。軸索ミトコンドリア減少ショウジョウバエは加齢に伴う運動機能の低下が若齢から始まり、寿命が短縮する(6)。
さらに近年、このミトコンドリア輸送の低下は、タンパク質恒常性を破綻させることによって老化を促進することがわかってきた。通常の老化によっても、細胞内のタンパク質分解系が低下し、本来分解されるべき不良タンパク質(ユビキチン化タンパク質)が脳内に蓄積する。しかし、ミトコンドリアを軸索から減少させると、タンパク質分解系の低下と不良タンパク質の蓄積が若齢から見られた(6)。
また、ミトコンドリアを軸索から減少させると、アルツハイマー病などの脳で神経細胞死を引き起こすタウタンパク質の毒性を増加させる。タウは通常、神経軸索の微小管の安定性を制御しているが、疾患脳では過剰にリン酸化され、細胞質に蓄積し凝集する。miltonのノックダウンによりタウのリン酸化が増加し、タウによる軸索変性が劇的に増加した(10)。すなわち、軸索でのミトコンドリア減少は、軸索での異常タンパク質に対する脆弱性を増加させるのである。
これらより、能動輸送によるミトコンドリアの局在制御が神経細胞のタンパク質恒常性の維持に必須であり、またその破綻が神経細胞の老化と、加齢による神経変性疾患のリスク増加の原因であることが考えられる。
ミトコンドリア異常の下流を標的とした治療戦略
ミトコンドリアの機能や輸送が低下しても、その不全をバイパスすることは可能だろうか?それらの戦略を組み合わせれば、神経細胞の老化を防げる可能性がある。
1.ミトコンドリア複合体II
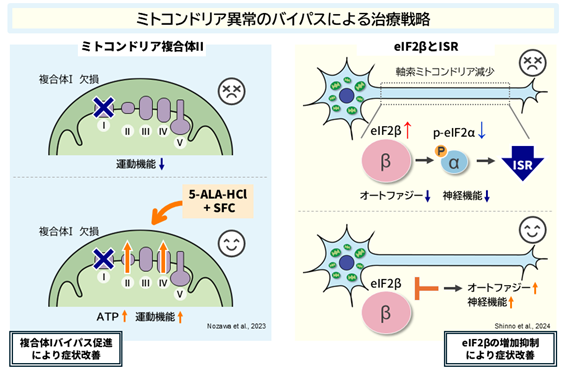
ミトコンドリア呼吸鎖複合体Iの欠損は小児のミトコンドリア病の原因として最も多い。しかし複合体Iをバイパスし、複合体II以降でATPを作る経路を活性化することで、その欠損を補い症状を緩和できる可能性がショウジョウバエモデルでの実験から明らかになった(11)。複合体I欠損による難病リー脳症のモデルショウジョウバエでは、ATP量が低下し、運動機能が障害され、寿命が短縮する。このショウジョウバエに5-アミノレブリン酸塩酸塩(5-ALA-HCl)とクエン酸第一鉄ナトリウム(SFC)を摂取させると、複合体Iは欠乏したまま複合体II, IVの活性が上昇し、ATPの増加と運動機能の改善がみられた(11)。複合体Iの減少はミトコンドリア病だけでなく、老化や多くの神経変性疾患で見られることから、複合体Iのバイパスがそれらの治療戦略になる可能性がある。
2.統合的ストレス応答(ISR)
翻訳開始因子eIF2はα, β, γからなる。eIF2複合体は通常、全般的な翻訳の開始に必須であるが、ストレス状態になるとeIF2αがリン酸化されることで全般的な翻訳を停止し、統合的ストレス応答(ISR)に則した翻訳へとスイッチングする。ISRはストレスに対して細胞を保護するが、一方でその過剰な活性化も神経細胞にダメージを与える可能性があり、その調節機構は神経細胞の維持に必須である(12)。私たちは軸索ミトコンドリア減少により、オートファジーによるタンパク質分解が低下するが、その原因がeIF2βの増加であることを突き止めた。加えて、軸索ミトコンドリア減少ショウジョウバエの脳内ではeIF2αのリン酸化が減少し、ISRが低下していた。興味深いことに、ミトコンドリアの軸索輸送が低下している状態でもeIF2βを減少させることで、神経機能の低下を抑制することができた(6)。加齢依存的なミトコンドリアの軸索輸送低下による神経細胞の老化に対し、eIF2βが標的になる可能性がある。
おわりに
老化とミトコンドリアの関係については、これまでATP産生や活性酸素の産生など呼吸鎖機能に注目した研究が主体であった。私たちを含め多くの研究グループの結果から、ミトコンドリアの機能活性化が神経変性疾患の治療標的になることは明らかである。一方で、軸索のミトコンドリアが減少することでオートファジーやISRに異常をきたすことは、ミトコンドリアの機能だけではなく局在そのものも重要であり、細胞内シグナル伝達のハブセンターになっていることを示唆している。今後、ミトコンドリアの局在や他の細胞内小器官との関係、細胞間コミュニケーションなど、加齢に伴うミトコンドリアの変化が脳内環境に与える影響を包括的に捉えていくことで、新たな治療ターゲットの発見が期待される。ミトコンドリアはまだまだ多くの謎を秘めており、その謎の中には老化や神経変性疾患の治療法の開発の鍵が眠っているかもしれない。
真野 叶子 博士(理学)
2018年北里大学理学部卒業。2024年東京都立大学大学院理学研究科修了、現在は名古屋大学 環境医学研究所 病態神経科学分野 特任助教。ミトコンドリアを興味の中心として、神経変性疾患や老化の研究をしています。

安藤 香奈絵 博士(薬学)
1996年東京大学薬学部卒業。同大学院薬学系研究科修了。米国コールドスプリングハーバー研究所を経て、2006年からトマスジェファーソン大学神経学科助教授、2014年東京都立大学理学部生命科学科准教授, 2025年から教授。研究室のメンバーとディスカッションを楽しみながら、広く脳の老化を研究しています。

■参考文献
- M. Oka et al., Increasing neuronal glucose uptake attenuates brain aging and promotes life span under dietary restriction in Drosophila. iScience 24, 101979 (2021).
- A. Ikeda et al., Mutations in CHCHD2 cause alpha-synuclein aggregation. Hum Mol Genet 28, 3895-3911 (2019).
- L. D. Goodman et al., Tau is required for glial lipid droplet formation and resistance to neuronal oxidative stress. Nat Neurosci 27, 1918-1933 (2024).
- I. Ralhan, C. L. Chang, J. Lippincott-Schwartz, M. S. Ioannou, Lipid droplets in the nervous system. J Cell Biol 220 (2021).
- L. Ruan et al., Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 543, 443-446 (2017).
- Y. M. Kanako Shinno, Koichi M Iijima, Emiko Suzuki, Kanae Ando, Axonal distribution of mitochondria maintains neuronal autophagy during aging via eIF2β. eLife 13 (2024).
- E. E. Glater, L. J. Megeath, R. S. Stowers, T. L. Schwarz, Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol 173, 545-557 (2006).
- X. Wang, T. L. Schwarz, The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163-174 (2009).
- A. Vagnoni, S. L. Bullock, A cAMP/PKA/Kinesin-1 Axis Promotes the Axonal Transport of Mitochondria in Aging Drosophila Neurons. Curr Biol 28, 1265-1272 e1264 (2018).
- K. Iijima-Ando et al., Loss of axonal mitochondria promotes tau-mediated neurodegeneration and Alzheimer’s disease-related tau phosphorylation via PAR-1. PLoS Genet 8, e1002918 (2012).
- N. Nozawa et al., 5-Aminolevulinic acid bypasses mitochondrial complex I deficiency and corrects physiological dysfunctions in Drosophila. Hum Mol Genet 32, 2611-2622 (2023).
- N. Calakos, Z. F. Caffall, The integrated stress response pathway and neuromodulator signaling in the brain: lessons learned from dystonia. J Clin Invest 134 (2024).
掲載元:Lab.First 研究ナレッジ