
免疫応答は厳重に制御されたプロセスであり、この厳密な制御を逸脱すると病理現象につながります。最近の研究では、免疫応答におけるメインシステムに加えて、免疫機能におけるイオンチャネルの重要な役割が明らかになってきています。ここではKV1.3およびKCa3.1(IKCa1)K+チャネルとプリン作動性受容体P2X7が、免疫応答の重要な要素の制御に大きくに関与していることについて述べていきます。
Noemí Bronstein-Sitton, Ph.D.
Cosmo Bio would like to acknowledge and thank the Alomone Labs for providing “Regulating the Immune Response” information presented here.
イオンチャンネルの意外な役割
一見しただけではわかりませんが、免疫系と中枢神経系(CNS;Central Nervous System)にはいくつかの類似点があります。どちらのシステムも複雑で動的な入力信号を処理し、適切かつタイムリーな反応を生み出していきます。そしてどちらのシステムも、応答が入力シグナルに確実に一致するように、またそれによって潜在的な損傷を防ぐように、絶妙に複雑なメカニズムを駆使しています。したがって現在、免疫学分野では、抗原提示細胞(APC;Antigen Presenting Cell)とTリンパ球細胞との密接な出会いと相互作用のことは、「シナプス」(あるいは「免疫学的シナプス」)という言葉を用いて説明されています。
しかし、免疫系の細胞はもともと「電気的に非興奮性」、つまり中枢神経系細胞の特徴である電位依存性イオンチャネルによって生成される活動電位を持たない細胞として分類されていました。ところが現在では免疫系細胞は電位依存性イオンチャネルや、その他の種類のイオンチャネルを数多く発現していることが明らかになっています1。イオンチャネルは中枢神経系と同様、免疫反応の制御においても重要な役割を果たしている可能性がでてきました。
T細胞活性化の調節
KV1.3とKCa3.1チャネルはT細胞の活性化に重要な役割を果たしています。KV1.3は電位依存性K+チャネルのShakerファミリーに属しています。このチャネルはTリンパ球・Bリンパ球・マクロファージ・ナチュラルキラー(NK)細胞など、造血系由来の様々な細胞集団に発現しています2。 Tリンパ球の機能的チャネルは4つのKV1.3サブユニットから構成され、静止細胞の膜電位を維持する役割を担っています。KCa3.1チャネル(IKCa1またはSK4とも呼ばれる)は、中程度のコンダクタンスを持つCa2+依存性K+チャネルです。このチャネルは電位感受性ではなく、μmolに満たない濃度の細胞内Ca2+で開口します。チャネルはそのC末端を介してCa2+センサーであるカルモジュリンと強固に結合しています3。KCa3.1は静止T細胞では低レベルで発現していますが、T細胞が完全に活性化されると劇的に発現量が増加します4,5。
特定の病原体に対する適応免疫応答は、Tヘルパー(TH)リンパ球が独自のT細胞レセプター(TCR)を介して関与または活性化されることで起こります(図1参照)。活性化されたT細胞は増殖し、特定の病原体に対処するのに最適な細胞サブセット(またはエフェクター)に分化します。侵入してきた病原体によって引き起こされる免疫反応の種類は、主に細胞内病原体やウイルスに対する細胞介在性(Th1とも呼ばれる)反応と、主に細菌や寄生虫に対する抗体依存性(またはTh2)反応(抗体産生Bリンパ球による)に分類されます。さらにその後、活性化されたT細胞はメモリー細胞に分化し、同じ病原体による再感染を予防するようになります。このようにAPCとT細胞との最初の出会いは、免疫反応の結果にとって極めて重要なのです。
T細胞が下す「決断」、すなわち反応するかどうか、どのように反応するかは、抗原の性質・APCとの遭遇期間・一般的な微小環境(サイトカイン、ケモカインなど)など、多くの因子によって制御される非常に複雑なプロセスです。まさにこの重要なプロセスにおいて、KV1.3とKCa3.1が関与していると考えられています。T細胞活性化におけるK+チャネルの関与を理解するための現在のモデルは、これらのチャネルがT細胞増殖に不可欠な細胞内Ca2+シグナルを間接的に制御していることを示唆しています。
抗原によるTCRが活性化に続く最も早い段階では、LckやPLCγ1などのタンパク質がチロシンリン酸化されます。後者はイノシトール1,4,5-三リン酸(IP3)の産生を通じて細胞内貯蔵物からのCa2+放出を誘導し、続いて細胞外空間からCa2+が持続的に流入します。活性化T細胞が増殖するためには、細胞内Ca2+濃度の上昇が比較的長時間(数時間)維持されなければならないことはよく知られています。細胞外Ca2+はその電気的駆動力に基づいて細胞内に入り込み、細胞膜を脱分極させます。これはKV1.3チャネルを活性化し、K+の流出を介して負の膜電位が維持され、その結果、継続的なCa2+の流入が可能になります。前述のように、T細胞の活性化はKCa3.1チャネルの発現を亢進し、KCa3.1チャネルは増加した細胞質Ca2+に反応して開口するため、膜電位をさらに過分極させます6,7。
まとめると、KV1.3とKCa3.1の両方が協調的に作用することで、細胞内Ca2+の持続的な上昇が可能になり、Ca2+依存的なカルシニューリンと転写因子NF-ATの活性化が可能になります。このモデルと一致して、CharybdotoxinやMargatoxinのようなK+チャネルのブロッカーは、抗原依存性のT細胞活性化とIL-2分泌を効果的に阻害していきます8,9,10。さらにKCa3.1の特異的阻害剤は、以前に活性化されたT細胞の活性化を阻害することが可能でした。これもまた活性化後のこれらのチャネルの発現増加が、その後の抗原チャレンジで重要な役割を果たすというモデルと一致しています11。
さらに、KV1.3チャネル阻害剤は、遅延型過敏症や外来抗原に対する抗体反応など、in vivoにおいてT細胞を介する免疫反応を阻害することが示唆されました12。これらの結果から、K+チャネルブロッカーは効果的な免疫抑制剤として使用できるのではないかという考えが生まれました。例えば神経炎症は複数の神経疾患の主な原因であり、ミクログリアの活性化によって引き起こされます。選択的自然免疫モデルマウスでは、Kv1.3はミクログリアの炎症性活性化に必要であることから、Kv1.3ブロッカーはミクログリアを介した神経毒性が病因に関与している場合、貴重な治療薬の候補となる可能性があります13。
免疫反応におけるK+チャネル全般の関与、特にKV1.3とKCa3.1の関与についてはここ数年で大きな進展がありましたが、まだ解明されていないことも多いようです。例えばある研究では主要なKVチャネルのオリゴマー組成は、機能的なリンパ球集団によって異なる可能性が示唆されました。ほかにもアネルギー化したT細胞(刺激を受けても増殖できない生細胞)は、KV1.3とともにKV1.2、KV1.1、またはKV1.6を発現している可能性があり、このことは、応答するT細胞が受ける生理的・機能的変化がKVチャネルのサブユニット組成および/または機能の変化と並行している可能性を示しています14。その証拠にT細胞を低酸素状態(酸素の利用可能性が低い状態)に曝露すると、KV1.3チャネルがタンパク質レベルでダウンレギュレートされるということが報告されており、この効果は、低酸素状態下での T 細胞増殖の抑制を説明できる可能性があります15。
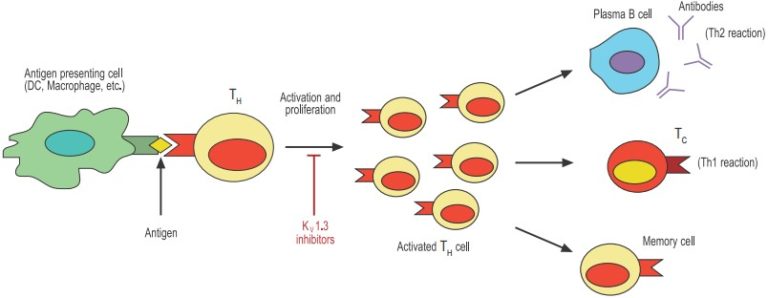
樹状細胞(DC)やマクロファージなどの抗原提示細胞は、細菌などの侵入病原体を貪食・消化する。病原体の選択されたペプチド(抗原)は、Tヘルパー(TH)細胞に「提示」される。適切なT細胞レセプターを持つTHだけが増殖し、活性化される。活性化されたTHは、血漿細胞から分泌される抗体やT細胞傷害性細胞(TC)などのエフェクター機構を刺激し、侵入してきた病原体を封じ込める。最後に、活性化されたTHはアポトーシスによって死滅するか、長寿命のメモリー細胞へと分化する。
炎症の制御:P2X7の中心的役割
P2X7受容体は、細胞外ATPに応答して開口するligand-gated型のイオンチャネルの大きなファミリーのメンバーです。このファミリーは7つのメンバー(P2X1~7)で構成されており、アミノ酸配列において40~50%の同一性を持ちます。すべてのP2X受容体が活性化されると、小さな陽イオン(Na+、K+、Ca2+)に対する膜透過性が増しますが、P2X7受容体を持続的に刺激すると低分子有機陽イオンが透過できる大きな膜孔が形成されます16。機能的なP2Xチャネルは多量体ですが、他のP2Xサブユニットとは対照的にP2X7はヘテロマーチャネルを形成することができません16。P2X7レセプターのもう一つの特異な特徴は、活性化するのに非常に高いATP濃度(mMの範囲)を必要とすることです。このことからP2X7受容体には別の内因性リガンドが存在するのではないかという推測がなされました。
P2X7受容体は造血系全体に広く発現しています。このレセプターはT細胞・B細胞・樹状細胞(DC)・マクロファージ・マスト細胞に存在し、いくつかの生理的活動に関与しています。炎症は喘息やアテローム性動脈硬化症のような病的状態の文脈で語られることが多いのですが、実際には組織の傷害や感染に対処するための複雑なメカニズムの重要な構成要素です。炎症は多くの場合、組織の傷害(例えば細菌の侵入によって引き起こされたものなど)が、好中球・マスト細胞・樹状細胞・マクロファージなどのさまざまな細胞集団を動員する化学的シグナルの分泌を開始するときに始まります。これらの細胞は防御の第一線を構成し、一方では貪食作用(マクロファージや好中球)によって侵入した細菌を攻撃し、他方では病原体の永久的な根絶を担う細胞(T細胞)へとつなぎます。炎症過程に関与するすべての細胞に発現しているP2X7受容体は、炎症制御において、重要な役割を担っていることが示唆されています(図2参照)。
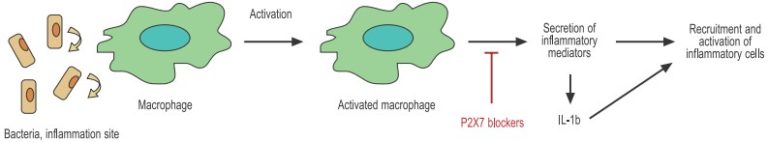
細菌は細胞膜の特異的受容体を通してマクロファージを活性化する。活性化されたマクロファージは、ケモカイン、サイトカインなどの炎症メディエーターを大量に分泌し、他の免疫細胞のリクルートと活性化を助ける。
免疫系におけるP2X7の最も驚くべき役割の一つとして、生物学的に活性なインターロイキン1β(IL-1β)の分泌における重要な役割が挙げられます。IL-1βは、メタロプロテアーゼのアップレギュレーションからIL-6の分泌に至るまで、さまざまなタイプの細胞から多種多様な炎症反応を引き起こす能力を持つことから、しばしば主要な炎症性サイトカインとみなされています17。IL-1βは、例えば感染部位に存在する細菌性LPS(Lipopolysaccharides: リポポリサッカライド)によって活性化されたマクロファージによって豊富に発現します。しかし、活性化されたマクロファージは、不活性なプロポリペプチド型のみを産生し、活性になる前にカスパーゼ-1によって切断する必要があります。驚くべきことに活性化マクロファージがカスパーゼ-1を活性化し、成熟IL-1βを産生するためには(LPSとは無関係な)第二の刺激が必要になります。このような状況で作用する最も強力なシグナルのひとつが、P2X7レセプターを介した細胞外ATPです18。
ATPを介したIL-1βの成熟におけるP2X7の中心的役割は、P2X7ノックアウトマウスから単離されたマクロファージで証明さています。これらの細胞はLPSに反応して多量の pro IL-1βを産生したにもかかわらず、成熟したIL-1βを産生することができませんでした19。免疫応答におけるP2X7のその他の機能としては、白血球やリンパ球におけるL-セレクチンの切断・ATPを介した増殖とアポトーシス・細胞内細菌の死滅・NF- Bの核への移行・単球やマクロファージにおけるシクロオキシゲナーゼ-2(COX-2)のアップレギュレーションといった細胞内炎症関連シグナル伝達経路の活性化などが知られています20,21,22。実際にP2X7を欠損させたマウス(P2X7ノックアウトマウス)は、炎症性関節炎のマウスモデルにおいて疾患の重症度が著しく低下したことから、P2X7が抗炎症薬の魅力的なターゲットになりうることが示唆されています23。
にもかかわらず、P2X7を活性化するためには非常に高濃度の細胞外ATPが必要であるという事実と、これらの知見を一致させることはこれまでは困難でした。
しかしある研究がその謎を解いてくれるかもしれません。その著者たちはニコチンアミドアデニンジヌクレオチド(NAD)が、ADPリボースとアクセプタータンパク質(この場合はP2X7)の結合を触媒する外部酵素であるADPリボース転移酵素2(ART2)の基質として機能することを示しました24。ART2を介したADP-リボシル化はP2X7を活性化し、ATP非存在下でCa2+の動員やL-セレクチンの切断など、既知の下流シグナル伝達を全て活性化することができます。これらの作用が起こるためには比較的低濃度(μmol範囲)の細胞外NADのみが必要でした。したがってP2X7の生理学的活性化の推定モデルは、炎症部位で死にゆく細胞の溶解によって放出された細胞外NAD(細胞外ATPとして)が、その部位に引き寄せられた炎症細胞の細胞膜上に発現したART2によって異化され、その結果、それらの細胞のP2X7チャネルを活性化するというものです。
その他の機能、その他のチャネル
これまで免疫反応の調節におけるKV1.3・KCa3.1・P2X7の関与について述べてきましたが、これは決して免疫反応におけるイオンチャネル全般の寄与を要約したものではありません。むしろすでにその関与が確認され、その機能が免疫反応の中心であるイオンチャネルは少なくないのです。
主な例はCa2+チャネルです。上述したように細胞内Ca2+濃度の上昇は、リンパ球(T細胞とB細胞の両方)や白血球(マクロファージ・好中球・マスト細胞)の活性化に極めて重要です。刺激に伴う細胞内貯蔵庫からのCa2+の放出はよく知られていますが、Ca2+レベルの上昇の大部分を担う細胞外培地からのCa2+の流入を媒介するチャネルの分子的な正体は、いまだに不明です。その不確かさは細胞膜Ca2+チャネルを説明するのに使われる用語にも表れています。Ca2+ release-activated Ca2+チャネル(CRAC)とstore-operated Ca2+チャネル(SOCC)という名称は同じ意味で使われていますが、両チャネルの生物物理学的特性には違いがあるかもしれません。CRACチャネルの分子的同一性の有力な候補はTRP遺伝子スーパーファミリーのメンバーです。T細胞ではTRPC3とTRPC6が研究されていますが、現在有力視されているのはTRPV6(CaT1)です25,26。
Cl–チャネルはリンパ球でも観察され、浸透圧ストレスの後に細胞の体積を減少させる働きをします1。さらにCl–チャネルのブロッカーは、リンパ球の活性化と増殖を阻害することが示されています27。CRACチャネルの場合と同様にCl–チャネルの分子的正体は明らかではありませんが、CLC3チャネルのmRNAはいくつかの造血細胞株で検出されています28。
最後になりますが、免疫系の細胞ではいくつかのイオンチャネルが同定されているものの、その機能はまだ十分に研究されていません。このグループにはリンパ球の上皮性Na+チャネル(ENaC)29、好中球の water チャネルのアクアポリン930・Bリンパ球のニコチン性アセチルコリン受容体31,32などが含まれます。
今後数年のうちに免疫反応におけるイオンチャンネルの新たな、そしておそらくはそれほど予期していなかった役割がさらなる研究によって明らかになることは間違いありません。
■参考文献
- Feske, S. et al. (2018) Annu. Rev. Immunol. 33, 291.
- Lewis, R.S. et al. (1995) Annu. Rev. Immunol. 13, 623.
- Logsdon, N.J. et al. (1997) J. Biol. Chem. 272, 32723.
- Ghanshani, S. et al. (2000) J. Biol. Chem. 275, 37137.
- Khanna, R. et al. (1999) J. Biol. Chem. 274, 14838.
- Cahalan, M.D. et al. (1997) Curr. Opin. Biotech. 8, 748.
- Matko, J. (2003) Trends Pharmacol. 24, 385.
- Lin, S.C. et al. (1993) J. Exp. Med. 177, 637.
- Leonard, R. J. et al. (1992) Proc. Natl. Acad. Sci. USA 89, 10094.
- Kerschbaum, H.H. et al. (1997) J. Immunol. 159, 1628.
- Wulff, H. et al. (2000) Proc. Natl. Acad. Sci. USA 97, 8151.
- Koo, G.C. et al. (1997) J. Immunol. 158, 5120.
- Lucente, J. et al. (2019) Glia 66, 1881.
- Liu, Q.H. et al. (2002) J. Exp. Med. 196, 897.
- Conforti, L. et al. (2003) J. Immunol. 170, 695.
- North, A.D. et al. (2002) Physiol. Rev. 82, 1013.
- Bent, R. et al. (2018) Int. J. Mol. Sci. 19, 2155.
- Perregaux, D.G. et al. (2000) J. Immunol. 165, 4615.
- Solle, M. et al. (2001) J. Biol. Chem. 276, 125.
- Fairbairn, I.P. et al. (2001) J. Immunol. 167, 3300.
- Budagian, V. et al. (2003) J. Biol. Chem. 278, 1549.
- Jamieson, G.P. et al. (1996) J. Cell Physiol. 166, 637.
- Labasi, J.M. et al. (2002) J. Immunol. 168, 6436.
- Seman, M. et al. (2003) Immunity 19, 571.
- Grafton, G. et al. (2001) Immunology 104, 119.
- Vassilieva, I. O. et al. (2013) J. Membr. Biol. 246, 131.
- Phipps, D.J. et al. (1996) Cell Signaling 8, 141.
- Jiang, B. et al. (2002) Life Sciences 70, 1383.
- Bubien, J.K. et al. (2001) J. Biol. Chem. 276, 8557.
- Loitto, V.M. et al. (2002) J. Leukoc. Biol. 71, 212.
- Skok, M.V. et al. (2003) Mol. Pharmacol. 64, 885.
- Fujii, T. et al. (2017) J. Pharmacol. Sci. 134, 1.
メーカー名:Alomone Labs
掲載元:Alomone Labs Article ;Regulating the Immune Response